CHLOROACETALDEHYDE
| Method no.: | 76 |
| Matrix: | Air |
| Target concentration: | 1.0 ppm (3.21 mg/m3) (OSHA ceiling PEL) |
| Procedure: | Air samples are collected by drawing known volumes of air
through |
| Recommended air volume and sampling rate: |
7.5 L at 0.5 L/min |
| Reliable quantitation limit: | 21.3 ppb (68.4 µg/m3) |
| Standard error of estimate at the target concentration: (Section 4.7.) |
7.67% |
| Special requirement: | Store collected samples in a freezer upon receipt at the laboratory. |
| Status of method: | Evaluated method. This method has been subjected to the established evaluation procedures of the Organic Methods Evaluation Branch. |
| Date: April 1989 | Chemist: Warren Hendricks |
OSHA Analytical Laboratory
Salt Lake City, Utah
1. General Discussion
- 1.1. Background
- 1.1.1. History
This method is similar to NIOSH Method S11 (Ref. 5.1.) for chloroacetaldehyde. The sampling rate was increased to better monitor ceiling exposures. Some of the recommended analytical parameters were changed to be consistent with OSHA analytical practices and techniques.
Chloroacetaldehyde can exist in combinations of four forms: the monomer, the monomer hydrate, the dimer hydrate and the cyclic trimer. The monomer and dimer hydrates are formed instantly when the anhydrous monomer is added to water. The cyclic trimer is formed from the anhydrous monomer upon standing. The cyclic trimer is only slightly soluble in water, but the monomer and dimer hydrates are formed when the cyclic trimer is heated in water. The anhydrous monomer is obtained by cracking the cyclic trimer. (Ref. 5.2.)
Commercial aqueous chloroacetaldehyde solution contains a 50/50 mixture of the two hydrates. When the aqueous mixture is.analyzed by GC, only a single peak at the same retention time as chloroacetaldehyde monomer is observed. The two hydrates are dehydrated and converted to the monomer during GC analysis. (Ref. 5.2.)
This method, like the NIOSH method, cannot distinguish between chloroacetaldehyde monomer and its monomer and dimer hydrates. Considering the ubiquitous nature of water in the environment, this issue is probably meaningless because chloroacetaldehyde monomer reacts instantly with water to form its monomer and dimer hydrates.
Chloroacetaldehyde, unlike some aldehydes, is sufficiently stable to permit its direct collection on silica gel and subsequent storage before analysis. Collected samples should be stored in a freezer upon receipt at the laboratory to prevent excessive migration from the front section to the backup section of the sampling tube.
1.1.2. Toxic effects (this section is for information only and should not be taken as the basis of OSHA policy.)
Chloroacecetaldehyde vapor is a severe irritant of the eyes, skin and mucous membranes. Direct contact with a 40% aqueous solution of chloroacetaldehyde can cause severe eye and skin burns. Chloracetaldehyde was found to be acutely toxic to rats, mice, rabbits, and guinea pigs following various routes of administration which included oral, intraperitoneal, and skin applications. (Ref. 5.3.)
Chloroacetaldehyde has been reported to be mutagenic by the Ames test. Hundreds of known carcinogens and noncarcinogens have been subjected to this test and about 85% of the known carcinogens were found to be mutagenic while less than 10% of the noncarcinogens gave a positive test. The fact that chloroacetaldehyde is a mutagen is of special interest because chloroacetaldehyde is a possible metabolic product of the human carcinogen vinyl chloride. It has been suggested that a metabolite of vinyl chloride may be the active carcinogenic form of the chemical. Chloroacetaldehyde is also a possible metabolic product of ethylene dichloride, ethylene chlorohydrin, and cyclophosphamide. (Ref. 5.4.)
1.1.3. Workplace exposure
The following are some common uses of chloroacetaldehyde during
which exposure may occur: (1) in the production of
Chloroacetaldehyde may be inadvertently released and exposure may occur when chloroacetaldehyde diethyl acetal is used in acid media during chemical syntheses (Ref. 5.5.).
1.1.4. Physical properties and other descriptive information (Ref. 5.3. unless otherwise indicated)
| CAS no.: | 107-20-0 |
| molecular weight: | 78.50 |
| synonyms: | |
| formula: | ClCH2CHO |
| Chloroacetaldehyde is combustible and forms an
insoluble hemihydrate at aqueous concentrations above 50%.
The following data are for a 40% aqueous solution: | |
| boiling point: | 90 to 100°C |
| vapor pressure: | 13 kPa (100 mm Hg) at 20°C |
| flash point: | 190°F (closed cup) |
| solubility: | water, acetone and methanol |
| The analyte air concentrations throughout this method are
based on the recommended sampling and analytical parameters. Air
concentrations listed in ppm are referenced to |
1.2. Limit defining parameters
- 1.2.1. Detection limit of the analytical procedure
The detection limit of the analytical procedure is 17.1 pg per injection. This is the amount of analyte which gave a chloroacetaldehyde peak whose height was about 5 times the height of a nearby trace contaminant peak. (Section 4.1.)
1.2.2. Detection limit of the overall procedure
The detection limit of the overall procedure is 0.513 µg per sample (21.3 ppb or 68.4 µg/m3). This is the amount of analyte spiked on the sampling device which allows recovery of an amount equivalent to the detection limit of the analytical procedure. (Section 4.2.)
1.2.3. Reliable quantitation limit
The reliable quantitation limit is 0.513 µg per sample (21.3 ppb or 68.4 µg/m3). This is the smallest amount of chloroacetaldehyde spiked on the sampling device which can be quantitated within the requirements of a recovery of at least 75% and a precision (±1.96 SD) of ±25% or better. (Section 4.3.)
The reliable quantitation limit and detection limits reported in the method are based upon optimization of the instrument for the smallest possible amount of the analyte. When the target concentration of the analyte is exceptionally higher than these limits, they may not be attainable at the routine operating parameters.
1.2.4. Instrument response to the analyte
The instrument response over the concentration range of 0.5 to 2 times the target concentration is linear. (Section 4.4.)
1.2.5. Recovery
The recovery of chloroacetaldehyde from samples used in a 17-day storage test remained above 90.4% when the samples were stored in the dark at about 22°C. (Section 4.5.) The recovery of an analyte from the collection medium during storage must be 75% or greater.
1.2.6. Precision (analytical procedure only)
The pooled coefficient of variation obtained from replicate injections of analytical standards at 0.5, 1, and 2 times the target concentration is 0.0063. (Section 4.6.)
1.2.7. Precision (overall procedure)
The precision at the 95% confidence level for the refrigerated 17-day storage test is ±15.0% (Section 4.7.). This includes an additional ±5% for sampling error. The overall procedure must provide results at the target concentration that are ±25% or better at the 95% confidence level.
1.2.8. Reproducibility
Six samples, spiked with chloroacetaldehyde, and a draft copy of this procedure were given to a chemist unassociated with this evaluation. The samples were analyzed after 11 days of storage at about 5°C. No individual sample result deviated from its theoretical value by more than the precision reported in Section 1.2.7. (Section 4.8.)
1.3. Advantage
This sampling and analytical method provides a simple and convenient means to monitor occupational exposure to chloroacetaldehyde.
1.4. Disadvantage
None
2. Sampling Procedure
- 2.1. Apparatus
- 2.1.1. Samples are collected by use of a personal sampling pump
that can be calibrated to within ±5% of the recommended flow rate
with the sampling device in line.
2.1.2. Samples are collected on 6-mm i.d. × 8-mm o.d. ×
2.2. Reagents
No sampling reagents are required.
2.3. Sampling technique
- 2.3.1. Break open both ends of the sampling tube so that the
holes in the tube ends are at least
2.3.2. Remove the sampling device after sampling for the appropriate time and then seal the tube with plastic end caps. Wrap the tube lengthwise with an official OSHA seal (Form 21).
2.3.3. Submit at least one blank sample with each set of samples. Handle the blank in the same manner as the other samples with the exception of drawing air through it.
2.3.4. List any potential interferences on the sample data sheet.
2.4. Sampler capacity
Five-percent breakthrough occurred after sampling a controlled test
atmosphere containing 6.3 mg/m3
chloroacetaldehyde (2 times the OSHA PEL) for 33.6 min at 0.5 L/min.
At the end of this time 16.8 L of air had been sampled and 105.8 µg of
chloroacetaldehyde had been collected. Breakthrough data were obtained
by sampling the test atmosphere for increasing periods of time with
several of the recommended
2.5. Desorption efficiency
- 2.5.1. The average desorption efficiency for chloroacetaldehyde
from 520 mg of silica gel (SKC, Inc., Lot 300) over the range of 0.5
to 2 times the target concentration was 91.0%. (Section 4.10.)
2.5.2. Desorbed samples remain stable for at least 16 h. (Section 4.10.)
2.6. Recommended air volume and sampling rate
- 2.6.1 The recommended air volume is 7.5 L.
2.6.2. The recommended air sampling rate is 0.5 L/min.
2.6.3. When longer-term air samples are required, reduce the sampling rate but do not exceed the recommended air volume.
2.7. Interferences (sampling)
Report suspected interferences to the laboratory with submitted samples.
2.8. Safety precautions (sampling)
- 2.8.1. The sampling equipment should be attached to the worker
in such a manner that it will not interfere with work performance or
safety.
2.8.2. All safety practices that apply to the work area being sampled should be followed.
3. Analytical Procedure
- 3.1. Apparatus
- 3.1.1. A GC equipped with an electron capture detector (ECD). A
3.1.2. A GC column capable of separating chloroacetaldehyde from
potential interferences. A
3.1.3. An electronic integrator or other suitable means of
measuring detector response. A
3.1.4. Four- and
3.2. Reagents
- 3.2.1. Chloroacetaldehyde. Chloroacetaldehyde, nominally 50% in
water, was obtained from Aldrich Chemical Co. and used in this
evaluation. The actual concentration of this solution must be
determined as described in Section 4.12.
3.2.2. Acetonitrile, reagent grade or better. American B&J brand acetonitrile was used in this evaluation.
3.2.3. 1,1,2-Trichloroethane, reagent grade or better.
3.2.4. The desorbing solution is acetonitrile containing 10 µL/L
of
3.3. Standard preparation
- 3.3.1. The concentration of the aqueous chloroacetaldehyde
solution must be determined as described in Section 4.12.
3.3.2. Prepare stock standards by diluting known amounts of the aqueous chloroacetaldehyde solution with desorbing solution. Store the stock standards in a refrigerator. Prepare fresh stock standards every two weeks.
3.3.3. Prepare analytical standards by further diluting stock standards with desorbing solution. Prepare fresh analytical standards each day.
3.3.4. Prepare a sufficient number of standards to generate a calibration curve. Analytical standard concentrations must bracket sample concentrations. Prepare additional standards if necessary.
3.4. Sample preparation
- 3.4.1. Transfer the 520-mg front section of silica gel to a 4-mL
glass vial. Place the
3.4.2. Add 3.0 mL of desorbing solution to each vial.
3.4.4. Seal the vials with PTFE-lined caps and allow them to desorb for 1 h. Mix the contents of the vials vigorously by hand several times during the desorption time.
3.4.5. Transfer some of the desorbed sample to autosampler vials. This step is necessary only if an autosampler is used.
3.5. Analysis
- 3.5.1. Analytical conditions
| GC conditions | |
| temperatures: | 40°C (column) 250°C (injector) 300°C (detector) |
| temperature program: | hold initial temp 5.0 min, increase temp at
|
| column head pressure: | 31 kPa (hydrogen) |
| column flow rate: | 1.7 mL/min (hydrogen) |
| septum purge flow rate: | 4.8 mL/min (hydrogen) |
| split vent flow rate: | 15.7 mL/min (hydrogen) |
| detector make-up flow rate: | 70 mL/min (nitrogen) |
| injection volume: | 1.0 µL |
| split ratio: | 1 to 10 |
| column: | A 30-m × 0.32-mm i.d. |
| chromatogram: | Section 4.11. |
3.5.2. Measure detector response using a suitable method such as electronic integration.
3.5.3. Use an internal standard procedure to prepare a calibration curve using several freshly prepared standards over a range of concentrations. Bracket the samples with analytical standards.
3.6. Interferences (analytical)
- 3.6.1. Any compound having a similar retention time as
chloroacetaldehyde or the internal standard is a potential
interference. Generally, chromatographic conditions can be altered
to separate an interference.
3.6.2. Retention time on a single column is not proof of chemical identity. Confirmation by GC/MS is a useful means of identification.
3.7. Calculations
- 3.7.1. Prepare calibration curves from analytical standards by
plotting detector response for chloroacetaldehyde versus the
analytical standard concentrations (in terms of micrograms
chloroacetaldehyde per milliliter). Determine the
3.7.2 Determine the concentration, in micrograms of chloroacetaldehyde per milliliter, of a sample by comparing its detector response to the calibration curve. Perform blank corrections for each section before adding the results together. If any chloroacetaldehyde is found on the backup section, add that amount to the amount found on the front section.
3.7.3. The air concentration of chloroacetaldehyde can be expressed in mg/m3 by using the following equation:
mg/m3 = [(A)(B)] / [(C)(D)]
| where | A = | µg/mL of chloroacetaldehyde from Section 3.7.2. |
| B = | desorption volume (3.0 mL) | |
| C = | liters of air sampled | |
| D = | desorption efficiency decimal form (0.910) |
3.7.4. Convert chloroacetaldehyde results in mg/m3 to ppm using the following equation:
ppm = (mg/m3)(24.46) / (78.50)
| where | mg/m3 = | results from Section 3.7.3. |
| 24.46 = | molar volume at 760 mm Hg and 25°C | |
| 78.50 = | molecular weight of chloroacetaldehyde |
3.8. Safety precautions (analytical)
- 3.8.1. Avoid skin contact and inhalation of all chemicals.
3.8.2. Restrict the use of all chemicals to a fume hood.
3.8.3. Wear safety glasses in all laboratory areas.
4. Backup Data
- 4.1. Detection limit of the analytical procedure
The detection limit of the analytical procedure is 17.1 pg per
injection. It is based on a
4.2. Detection limit of the overall procedure
The detection limit of the overall procedure is 0.513 µg per sample (21.3 ppb or 68.4 µg/m3). The injection size recommended in the analytical procedure (1.0 µL with a 1 to 10 split) was used in the determination of the detection limit of the overall procedure. Six vials containing 520 mg of silica gel were each liquid spiked with 0.513 µg of chloroacetaldehyde. The samples were desorbed about 16 h after being spiked.
|
| ||
| sample number |
theoretical amount (µg) |
amount recovered (µg) |
|
| ||
| 1 | 0.513 | 0.483 |
| 2 | 0.513 | 0.472 |
| 3 | 0.513 | 0.493 |
| 4 | 0.513 | 0.452 |
| 5 | 0.513 | 0.463 |
| 6 | 0.513 | 0.473 |
|
| ||
4.3. Reliable quantitation limit data
The reliable quantitation limit is also 0.513 µg per sample (21.3 ppb or 68.4 µg/m3). The injection size recommended in the analytical procedure (1.0 µL with a 1 to 10 split) was used in the determination of the reliable quantitation limit. Six vials containing 520 mg of silica gel were each liquid spiked with 0.513 µg of chloroacetaldehyde. Because the recovery of chloroacetaldehyde from the spiked samples was greater than, 75%, and because the precision (1.96 SD) was less than ±25%, the detection limit of the overall procedure and reliable quantitation limit are the same.
|
| ||
| percent recovered |
statistics | |
|
| ||
| 94.2 | ||
| 92.0 | 92.1 | |
| 96.1 | SD = | ±2.83 |
| 88.1 | Precision = | (1.96)(±2.83) |
| 90.2 | = | ±5.5 |
| 92.2 | ||
|
| ||
4.4. Instrument response to chloroacetaldehyde
The instrument response to chloroacetaldehyde over the range of 0.5 to 2 times the target concentration is linear with a slope of 1405 area counts per microgram per milliliter. The response to chloroacetaldehyde was determined by multiple injections of standards. The data in Table 4.4. is presented graphically in Figure 4.4.
|
| |||
| × target conc. µg/sample |
0.5× 4.17 |
1× 8.33 |
2× 16.67 |
|
| |||
| area counts | 7578 | 14290 | 25268 |
| 7590 | 14245 | 25502 | |
| 7635 | 14350 | 25469 | |
| 7644 | 14298 | 25385 | |
| 7749 | 14269 | 25244 | |
| 7744 | 14351 | 25446 | |
| 7656.7 | 14300.5 | 25385.7 | |
|
| |||
4.5. Storage data
Thirty-six samples were generated by sampling a test atmosphere
containing 3.18 mg/m3 chloroacetaldehyde for
15 min at 0.5 L/min. The relative humidity of the atmosphere was 76%
at 24°C.
|
| ||||||||
| storage time (days) |
ambient (% recovery) |
refrigerated (% recovery) | ||||||
|
| ||||||||
| 0 | 88.6 | 87.9 | 89.0 | 91.6 | 98.6 | 101.6 | ||
| 0 | 91.6 | 98.6 | 101.6 | 88.6 | 87.9 | 89.0 | ||
| 3 | 91.0 | 97.6 | 89.7 | 95.0 | 94.6 | 105.4 | ||
| 8 | 90.9 | 100.4 | 97.2 | 97.7 | 102.1 | 106.2 | ||
| 10 | 88.0 | 88.5 | 86.5 | 93.4 | 95.0 | 98.3 | ||
| 14 | 86.5 | 86.5 | 94.7 | 94.2 | 105.4 | 89.8 | ||
| 17 | 87.3 | 90.1 | 98.4 | 96.3 | 89.8 | 100.0 | ||
|
| ||||||||
4.6. Precision (analytical method only)
The precision of the analytical procedure is defined as the pooled coefficient of variation determined from replicate injections of chloroacetaldehyde standards at 0.5, 1, and 2 times the PEL.
|
| |||
| × target conc. µg/sample |
0.5× 4.17 |
1× 8.33 |
2× 16.67 |
|
| |||
| SD1 | 74.07 | 42.93 | 107.73 |
| CV | 0.00967 | 0.00300 | 0.00424 |
|
| |||
| 1 standard deviation is in area counts | |||
4.7. Precision (overall procedure)
The precision of the overall procedure is determined from the storage data. The determination of the standard error of estimate (SEE) for a regression line plotted through the graphed storage data allows the inclusion of storage time as one of the factors affecting overall precision. The SEE is similar to the standard deviation except it is a measure of dispersion of data about a regression line instead of about a mean. It is determined with the following equation:
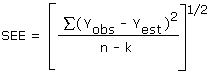
| where | |
| n = k = k = |
total no. of data points 2 for linear regression 3 for quadratic regression |
| Yobs =
Yest = |
observed % recovery at a given time estimated % recovery from the regression line at the same given time |
An additional ±5% for pump error is added to the SEE by the
addition of variances. The precision at the 95% confidence level is
obtained by multiplying the SEE (with pump error included) by 1.96
(the
4.8. Reproducibility data
Six samples, liquid spiked with chloroacetaldehyde, were given to a chemist unassociated with this study. The samples were analyzed after being stored for 11 days at about 5°C. No sample result had a percent deviation greater than the precision of the overall procedure, which was ±15.0%.
|
| |||
| µg spiked | µg recovered | % recovered | % deviation |
|
| |||
| 25.65 | 24.21 | 94.4 | -5.6 |
| 25.65 | 23.92 | 93.2 | -6.8 |
| 25.65 | 24.45 | 95.3 | -4.7 |
| 25.65 | 23.97 | 93.4 | -6.6 |
| 25.65 | 23.43 | 91.3 | -8.7 |
| 25.65 | 23.97 | 93.4 | -6.6 |
|
| |||
4.9. Sampler capacity
Five-percent breakthrough occurred after sampling a controlled test atmosphere containing 6.3 mg/m3 chloroacetaldehyde at 67% relative humidity and 27°C for 33.6 min at 0.5 L/min. At the end of this time 16.8 L of air had been sampled and 105.8 µg of chloroacetaldehyde had been collected. Breakthrough data were obtained by sampling the test atmosphere for increasing periods of time with several of the recommended two section silica gel tubes. Breakthrough was defined as the amount of chloroacetaldehyde found on the backup section relative to the total amount found on the entire tube. The results of the breakthrough test are also presented in Figure 4.9.
|
| |
| air volume (L) |
breakthrough (%) |
|
| |
| 7.14 | 0.0 |
| 7.26 | 0.0 |
| 14.13 | 0.67 |
| 14.75 | 2.41 |
| 21.95 | 17.64 |
| 23.48 | 22.25 |
|
| |
4.10. Desorption efficiency and stability of desorbed samples
- 4.10.1. Desorption efficiency
The desorption efficiency (DE) of chloroacetaldehyde was
determined by
|
| |||
| × target conc. µg/sample |
0.5× 12.5 |
1× 25.0 |
2× 50.0 |
|
| |||
| DE, % | 91.9 | 91.4 | 90.2 |
| 92.1 | 89.5 | 91.1 | |
| 93.2 | 90.1 | 92.3 | |
| 91.5 | 88.8 | 91.7 | |
| 91.5 | 89.8 | 91.2 | |
| 92.1 | 88.8 | 92.1 | |
| 92.0 | 89.7 | 91.4 | |
|
| |||
4.10.2. Stability of desorbed samples
The stability of desorbed samples was investigated by reanalyzing the "1×" target concentration desorption samples about 16 h after the original analysis. The samples were resealed immediately following the first analysis, stored at ambient temperature, and were reanalyzed with fresh standards. The average of the reanalyzed samples relative to the average of the original analysis was 100.8%.
|
| ||
| original result (%) |
reanalyzed result (%) |
reanalyzed relative to original (%) |
|
| ||
| 91.4 | 91.2 | 99.8 |
| 89.5 | 90.0 | 100.6 |
| 90.1 | 90.6 | 100.6 |
| 88.8 | 90.0 | 101.4 |
| 89.8 | 91.1 | 101.4 |
| 88.8 | 89.5 | 100.8 |
|
| ||
4.11. Chromatogram
A chromatogram at the target concentration is shown in Figure 4.11.
4.12. A procedure to determine chloroacetaldehyde by titration
This procedure was taken from Reference 5.1. It is based on the reaction of chloroacetaldehyde with hydrazine sulfate to convert chemically bound chlorine to chloride. Silver nitrate, in excess of the amount required to react with the liberated chloride, is added to the sample. The excess silver is titrated with ammonium thiocyanate using ferric ammonium sulfate as an indicator.
- 4.12.1. Apparatus
Miscellaneous glassware. A 25-mL burette, 250-mL Erlenmeyer flasks, 100-mL volumetric flask, weighing bottle, pipets, etc.
4.12.2. Reagents
- 4.12.2.1. Hydrazine sulfate, 99% A.C.S. reagent. Hydrazine
sulfate was purchased from Aldrich. Prepare a 2.5% (by weight)
solution in deionized water. CAUTION. Hydrazine sulfate is
a cancer suspect agent.
4.12.2.2. Ferric ammonium sulfate, 99% A.C.S. reagent. Ferric ammonium sulfate was purchased from Aldrich. Prepare a saturated solution in 1 M aqueous nitric acid.
4.12.2.3. Nitric acid, 69.0-71.0%. "Baker Analyzed" reagent grade was purchased from VWR Scientific. Prepare a 50/50 (v/v) solution in deionized water and boil until colorless to remove nitrous acid.
4.12.2.3. Ammonium thiocyanate, 0.100 N, volumetric solution. "Mallinckrodt Standard" grade 0.100 N solution was purchased from Baxter Scientific.
4.12.2.4. Silver nitrate, 0.1000 N, volumetric standard. A standardized 0.1002 N solution was purchased from Aldrich.
4.12.2.5. Nitrobenzene, reagent grade. "Baker Analyzed" reagent grade was purchased from VWR scientific.
4.12.3. Procedure
- 4.12.3.1. Weigh (using a weighing bottle) 1 mL of the 50%
chloroacetaldehyde (Section 3.2.1.) into a
4.12.3.2. Pipet two 5-mL aliquots of the diluted
chloroacetaldehyde solution into separate
4.12.3.3. Add 10.0 mL of the 0.1000 N silver nitrate solution, 5 mL of the 50/50 nitric acid solution and 2 mL of the saturated ferric ammonium sulfate solution to each flask. Swirl the contents of the flasks and then add 2 mL of nitrobenzene to each flask. The nitrobenzene serves to prevent the reaction of the precipitated silver chloride with ammonium thiocyanate.
4.12.3.4. Titrate the contents of each flask with 0.100 N ammonium thiocyanate solution to a permanent faint orange/brown endpoint.
4.12.4. Calculations
Calculate, using the following equation, the volumes of standard silver nitrate required to react with the chloride liberated from the sample (Vs) and also the amount present in the blank (Vb).
- Vs or Vb = 10-(A(B/C))
| where | A = | volume of ammonium thiocyanate required for titration in Section 4.12.3.4., mL |
| B = | normality of ammonium thiocyanate solution | |
| C = | normality of silver nitrate solution |
Calculate, using the following equation, the percent of chloroacetaldehyde (CAA) present in the original solution.
- CAA, % wt =
[(Vs-Vb)(C)(7.85)(20)]/D
| where | 7.85 = | chloroacetaldehyde equivalent factor |
| 20 = | dilution factor | |
| D = | weight from Section 4.12.3.1., g |
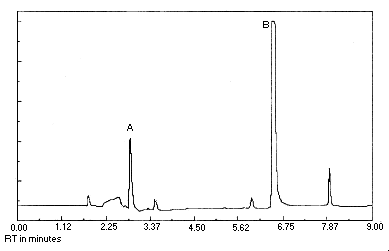
Figure 4.1. Detection limit of the analytical procedure. Peak identification was as follows: A, chloroacetaldehyde; B,
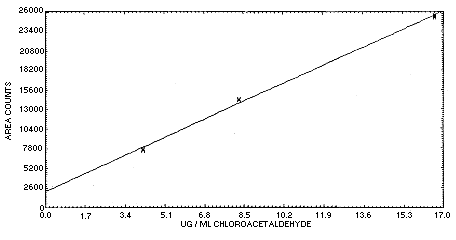
Figure 4.4. Chloracetaldehyde calibration curve.
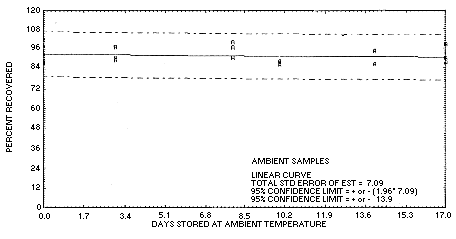
Figure 4.5.1. Ambient
temperature storage test for chloroacetaldehyde.
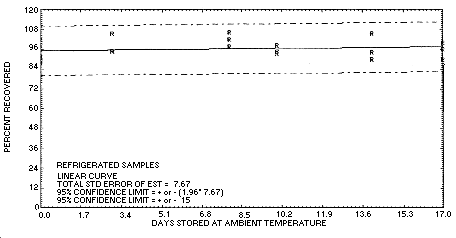
Figure 4.5.2. Refrigerated
temperature storage test for chloroacetaldehyde.
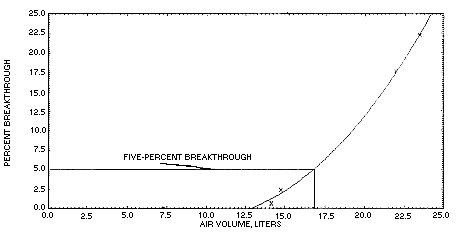
Figure 4.9. Sampler
capacity test.
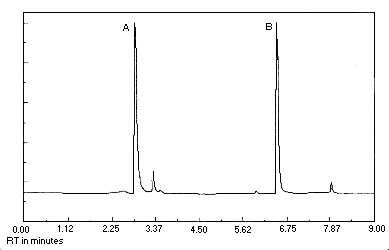
Figure 4.11. Chromatogram at the target concentration. Peak identification was as follows: A, chloroacetaldehyde; B,
5. References
- 5.1. "NIOSH Manual of Analytical Methods", 2nd ed.; U.S.
Department of Health and Human Services, Center for Disease Control,
NIOSH; Cincinnati, OH, 1979, Vol. 5, Method S11, DHEW (NIOSH) Publ.
No.
5.2. Elmore, J.D.; Wong, J.L.; Laumbach, A.D. and Streips, U.N.
Biochimica et Biophisica Acta, 1976, 442, pp
5.3. "Documentation of the Threshold Limit Values and Biological
Indices", 5th ed.; American Conference of Government Industrial
Hygienists (ACGIH): Cincinnati, ISBN:
5.4. McCann, J.; Simmon, V.; Streitwieser; Ames, B.N. Proc. Nat.
Acad. Sci. U.S.A., 1975, 8, pp
5.5. "NIOSH/OSHA Occupational Health Guidelines for Chemical
Hazards", U.S. Dept. of Health and Human Services, Public Health
Services, Center for Disease Control, NIOSH and U.S. Dept. of Labor,
OSHA: U.S. Government Printing Office Washington, DC, Jan 1981,
Chloroacetaldehyde, DHHS (NIOSH) Publ. No.
5.6. "The Merck Index", 10th. ed., Windholz, M. Ed., Merck & Co.: Rahway, NJ, 1983, p 296.